
受限于空间和时间分辨率,目前对原子级微观动态过程的理解尚存在局限性,这在一定程度上严重制约了催化剂调控技术的发展及其应用领域的扩展。面对这一挑战,研究团队致力于高性能敏感材料的设计与调控工作,旨在实现对环境污染物及体液电解质离子的高灵敏度和高选择性检测。目前,团队已研发了系列界面新结构和表征新方法,例如,设计Au25纳米簇负载在黑磷和氮掺杂石墨烯的“三明治结构”材料(AuCs/BP-NG),“再分散”效应实现了对Cr(VI)的高稳定高灵敏催化检测(Adv. Funct. Mater., 2022, 2209283);通过原位电化学电子自旋共振波谱技术,探究由氧空位引发的电子重排所促进的羟基自由基高效界面催化(Chem.Sci., 2023, 14, 2960);结合密度泛函理论和梯度提升回归方法,全面筛选过渡金属单原子高效催化As(III)的通用描述符(Anal. Chem., 2023, 95, 3666);借助计算机断层扫描技术提取界面材料几何特征模拟,阐明极低界面扩散电位下的双层电容转导机制(ACS Nano, 2024, 18, 12808)。
该研究中,研究人员结合了先进的原位同步辐射光谱技术和高精度的理论计算,实现了对双金属单原子催化剂在催化反应过程中瞬态结构的实时捕捉和解析。首先,通过高通量筛选确定了高效的双金属原子级电极界面,实现了Cu(II)和As(III)的并行电化学还原。原位X射线吸收精细结构(XAFS)光谱和配位场理论验证了双金属单原子体系中,由允许的d−d跃迁促进的Ni−Cu特定能级匹配,再现了电化学微观动态还原过程。进一步通过理论计算发现,Fe−As特定键合和最小势能(1.40 eV)决定步骤,这是源自关键中间体的主要s和p峰向高能轨道的线性偏移。此外,研究人员分别从动力学、热力学模型的收敛趋势和退火模拟的角度再现了自适应匹配的动态演化。
该研究中高通量计算方法的应用显著加速了催化剂候选材料的筛选过程,通过预测材料的催化性能和稳定性,缩小了实验验证的范围,从而大大提高了研究效率。瞬态反应机制研究则进一步揭示了催化过程中关键步骤的热/动力学特征,为优化催化剂性能提供了理论指导。另外,原位实验验证证实了理论预测的准确性,精细的表征手段也揭示了催化剂的结构-性能关系,为深入理解催化机制提供了坚实依据。该工作结合理论和实验的指导方法,不仅证实了高效双功能纳米材料理论预测的可行性,也为下一代多金属原子级催化剂的智能设计、理论筛选及催化机制原位探究提供了重要参考。
固体所博士后宋宗银为论文的第一作者,安光所刘文清院士、固体所黄行九研究员和中国科学院上海应用物理研究所李丽娜教授为通讯作者。该研究得到了国家重点研究开发计划、中国博士后创新人才支持计划、国家自然科学基金及中国科学院青年创新促进会等项目的资助。
文章链接:https://doi.org/10.1021/acs.nanolett.4c06524
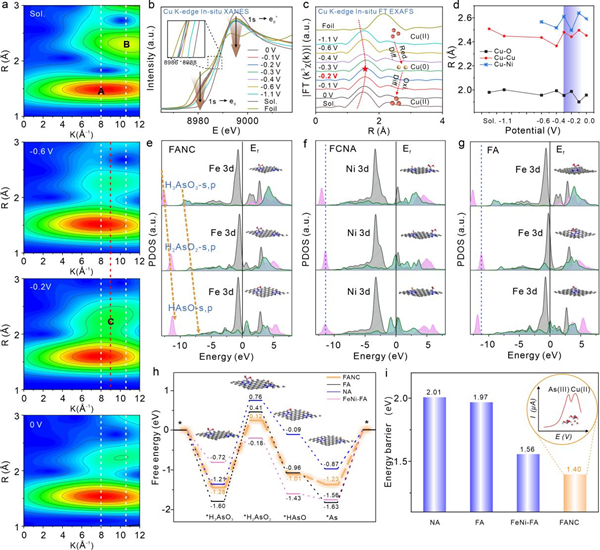
图1. 原位XAFS技术和DFT计算模拟联用探究电化学反应微观动态演变。

图2. 作为封面论文。
